热门搜索:分会介绍 | 会员名单 | 行业资讯

准确和灵敏地检测罕见突变,对于CRISPR基因组编辑技术的安全应用和癌症的早期诊断等一系列研究具有重要意义。虽然现有的高通量测序技术已经有了长足的进步,但是目前由于建库和测序过程中导致的错误还是在所难免,所以难以检测到大量样品中的罕见突变【1】。尤其对于罕见的复杂结构变异,还没有有效的检测和定量的方法。纳米孔测序(Nanopore sequencing)的长读长优势,有利于检测复杂的结构变异,但由于其自身的高错误率(~8%),很难用于罕见突变检测。近期研究表明CRISPR/Cas9基因编辑除了会在目标位点导致小范围的DNA序列突变,也有可能会产生长达数千碱基的DNA删除或其它结构变异【2】。这类之前未被重视的复杂的大范围变异引发了对CRISPR/Cas9基因编辑安全性的质疑。但由于其发生的频率通常较低且范围广,不容易被短读长的二代测序检测到,针对此类突变的研究一度陷入僵局。
2020年8月25日,沙特阿拉伯阿卜杜拉国王科技大学(KAUST)生物与环境科学工程学院的李墨(Mo Li)教授和北京大学生物医学前沿创新中心(BIOPIC)、北京未来基因诊断高精尖创新中心(ICG)的黄岩谊教授合作在Genome Biology上发表了题为Long-read individual-molecule sequencing reveals CRISPR-induced genetic heterogeneity in human ESCs的文章【3】。该研究提出了一种基于分子标签(unique molecular identifier, UMI)的单分子标记技术(targeted Individual DNA Molecule Sequencing, IDMseq),用于定量的检测样品中的罕见突变,包括单碱基突变以及大范围的复杂的结构变异。

研究团队利用分子标签定向的标记样品中目标基因的原始DNA分子,之后利用PCR的方式扩增标记到的分子用以测序。该技术配套的数据分析算法VAULT(variant analysis with unique molecular identifier for long-read technology)可以从测序结果中发现并提取分子标签,然后利用分子标签将测序的数据(read)分组。每个组都代表初始体系中的一个DNA分子,于是可以利用同一个组内的所有数据进行测序纠错,从而得到高精度的突变结果。该技术广泛适用于当前的主流高通量测序平台,包括Illumina, PacBio 和Nanopore。研究表明运用该技术后,高错误率的Nanopore测序可以提供媲美Illumina的罕见突变检测效果,准确的检测到了预设的1:100、1:1000、1:10000的罕见点突变并正确的报告出该突变的频率。
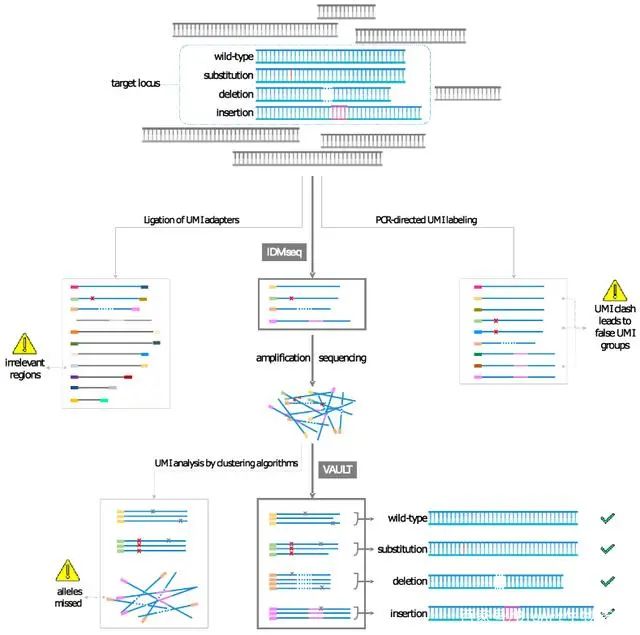
研究团队之后利用该技术检测CRISPR/Cas9基因编辑引起的突变。团队通过IDMseq技术对Cas9酶切位点附近的7-8kb范围进行测序。研究表明,在Cas9编辑后的人多显能胚胎干细胞(hESC)中, 2.8-5.4% 的DNA分子存在大删除突变。发现的最长删除的距离超过5.5kb。其中很多删除突变发生在相同的位点,表明可能存在这类突变的热区。除大范围的突变外,在基因编辑的区域DNA点突变数量增加了三倍。值得注意的是,这些点突变频率很低(<1%),因而很难被常规测序方法捕捉到。研究团队的这一系列发现,增加了对CRISPR/Cas9基因编辑安全性的认识,同时也为后续相关研究提供了方法支撑。
据悉,沙特阿拉伯阿卜杜拉国王科技大学(KAUST)生物与环境科学工程学院的毕重伟和王琳(现就职于吉林大学人兽共患病研究所)为本文的共同第一作者。沙特阿拉伯阿卜杜拉国王科技大学(KAUST)生物与环境科学工程学院的李墨(Mo Li)教授和北京大学的黄岩谊教授为本文的共同通讯作者。此研究也得到了阿卜杜拉国王科技大学计算生物学研究中心的高欣教授的支持。
邮政编码:200052 电话:021-63800152 传真:021-63800151 京ICP备15010734号-10 技术:网至普网站建设
 扫描下载
扫描下载 扫描关注
扫描关注 扫描关注
扫描关注 扫描关注
扫描关注 扫描关注
扫描关注