热门搜索:分会介绍 | 会员名单 | 行业资讯

PD-L1抗体试剂的审评挑战
免疫治疗自2013年被评为“突破性疗法”以来,美国、欧洲、日本等国家已批准了多个用于不同癌种的PD-1/PD-L1免疫药物,我国也于近期批准上市了PD-1免疫药物。随着PD-1/PD-L1免疫药物的广泛使用,如何应用免疫标志物选择合适的治疗人群成为免疫治疗面临的主要挑战之一。
多项大型临床研究显示,在肿瘤患者中,PD-1/PD-L1抑制剂治疗的获益程度与肿瘤患者PD-L1表达存在相关性。通过免疫组织化学的方法检测肿瘤患者的PD-L1表达是目前公认的最为有效的方法。对肿瘤患者的PD-L1表达水平进行检测,可以鉴别出哪些肿瘤患者适合使用PD-1/PD-L1抑制剂进行治疗。
目前世界各国针对PD-1/PD-L1抑制剂开发了多个基于免疫组织化学方法的检测试剂,用于评估PD-L1的表达情况,包含不同的抗体、克隆、平台、评分系统和临界值设定,分别作为各自药物的伴随诊断试剂。但4种PD-1/PD-L1抑制剂临床研究中应用的检测抗体(克隆号分别为28-8、22C3、SP142、SP263)均未在我国获得上市批准。目前,克隆号为SP263、SP142和22C3的抗体试剂在我国已进入注册申请过程,其中部分申报产品进入优先审查程序。
PD-L1抗体试剂的审评难点集中在临床试验部分。首先,PD-L1抗体试剂的判读方式与一般辅助诊断用途的免疫组织化学抗体试剂有所不同。PD-L1的表达情况是通过计算任何染色强度下肿瘤细胞和/或免疫细胞染色的百分比予以评定。然而,不同克隆的PD-L1表达判读标准以及阈值设定并不相同,阳性染色位置设定亦不相同,造成了PD-1/PD-L1抑制剂研究结果无法互相通用或横向比较。如何选择一致性研究中的对比试剂及合理进行平行判读结果的比较,是审评过程中需要考虑的问题。
另外,目前国内多家单中心初步研究数据显示,我国肺癌患者的PD-L1表达与国外研究稍有不同,不同病理类型癌种的PD-L1表达情况与国外存在差异。因此,在伴随诊断临床意义的验证中,如何合理接受境外临床试验数据也是审评过程中的挑战。
关于人类Brca1/2基因突变检测试剂注册审评概述
一、Brca1基因和Brca2基因概述
Brca1基因和Brca2基因都是常染色体显性遗传的肿瘤抑制基因,参与DNA的损伤修复和转录调控。研究表明,Brca1/2基因突变的肿瘤细胞对PARP抑制剂的敏感性显著高于正常细胞,通过抑制PARP-1的活性可使Brca1/2基因突变的肿瘤细胞发生合成致死。修饰酶PARP-1(Ⅰ型多聚ADP-核糖聚合酶,poly(ADP-ribose) polymerase 1)在DNA损伤修复和细胞凋亡中发挥重要作用,Brca1基因和Brca2基因发生突变的肿瘤细胞往往依赖PARP-1完成对DNA损伤的修复,因此PARP抑制剂通过抑制PARP-1的功能可靶向杀伤Brca1/2基因突变的肿瘤细胞。临床研究表明,Brca1基因或Brca2基因突变的卵巢癌和乳腺癌患者可以从PARP抑制剂奥拉帕尼和Rubraca等治疗中获益。
目前美国食品与药物管理局(FDA)和欧洲药品管理局(EMA)等已批准PARP抑制剂用于治疗Brca1基因或Brca2基因突变的卵巢癌患者。奥拉帕尼治疗乳腺癌的临床Ⅲ期研究于2017年宣布成功结束,达到临床设计的首要终点目标,结果显示可显著延长患者无进展生存期,2018年12月美国FDA批准奥拉帕尼用于携带Brca突变阳性的晚期卵巢癌患者一线含铂化疗完全或部分缓解患者的维持治疗。该药已于2018年8月在中国批准上市。
二、Brca1基因和Brca2基因检测试剂及审评难点
美国FDA陆续批准了Brca1/2突变基因检测产品上市。该类产品其共同特点是由美国FDA指定该类产品在单一检测地点进行检测。而目前国内部分专家并不完全认同该模式,认为国内临床机构的检测和判读能力不低于企业的检测和判读能力,且中美医疗环境及该类产品实际临床检测应用发展存在差异。部分临床机构配备相关人员,具有较强的检测和解读能力,但因Brca检测业务在中国开展较晚,只少数医院能够满足上述条件。部分临床机构目前不具备相关检测能力和/或解读能力,质量难以评价和控制。
开展基于NGS测序原理的Brca检测需要配备NGS实验室及相关试验人员及生物信息解读人员等,目前欧洲分子基因诊断质量联盟(European Molecular Genetics Quality Network,EMQN)和国家卫生和计划生育委员会病理质控评价中心(PQCC)会对Brca每年进行室间质评,国内部分检验机构也进行过Brca项目的室间质评。但目前对于NGS实验室还没有有效的体系规范和考核,对解读人员资质的认可也缺乏有效手段。
国内目前没有公认的人类遗传病数据库,数据对比主要依靠国外数据。如何让申请人的软件接入的数据库充分且合理,以及如果国内出现数据库,什么样的数据库是可接受的等问题,都是需要进一步探索的问题。
三、小结
通过前期调研和讨论,我部门认为该类产品的检测应用机构能力非常重要、检测结果解读较为复杂,以及目前中国人群遗传信息数据缺乏等问题,需要在其申请上市时对应用检测地点、产品质量、结果解读、产品监管等方面加强考虑。
考虑到该类产品有重要的临床意义,为了保障检测质量和结果解读的能力。在技术审评过程中,我们将考虑结合产品将其应用范围限制在一定检测范围内。如申请人相关的检测中心及一定范围内的临床机构。
Brca检测应用中,Brca遗传信息数据库是结果判断的重要依据。理想的遗传性疾病数据库应该做到公开、可溯源、不可篡改数据、符合伦理要求、有专人或团队负责数据的更新和维护等。鼓励国内大的遗传数据库按照上述原则进行建设,并逐步建立国内中国人群数据库。
综上,我们将积极探索我国该类产品的审评模式和上市相关问题。
水通道蛋白抗体检测试剂注册审评概述
水通道蛋白抗体(AQP4-IgG)检测对于视神经脊髓炎(NMO)诊断具有显著临床意义。目前我国尚无该类试剂上市,此种情况给临床NMO诊断带来了一定的困难。《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》明确提出:“支持罕见病治疗药品医疗器械研发,对境外已批准上市的罕见病治疗药品医疗器械,可附带条件批准上市。”为了支持和鼓励相关产品研发,国家药品监督管理局发布了《用于罕见病防治医疗器械注册审查指导原则》。作为技术审评机构,在相关产品审评过程中,会依据法规政策和技术文件,结合该类产品的特点,科学审评,优先审评,促进该类产品尽快用于临床,使罕见病患者受益。
视神经脊髓炎(NMO)是一种以视神经和脊髓受累为主的中枢神经系统炎性脱髓鞘疾病,是目前公认的神经系统自身免疫性疾病。NMO临床上多以严重的视神经炎和纵向延伸的长节段横贯性脊髓炎为特征表现,常于青壮年起病,女性居多,复发率及致残率高,可导致失明和瘫痪。NMO是一种罕见病,其在非白种(亚洲、拉丁美洲、非洲、西班牙裔和美国原住民)人群中更为易感,目前已列入我国《第一批罕见病目录》。
AQP4-IgG是视神经脊髓炎的特异性标记物,作为诊断指标被列入相关国际标准和国内的《中国视神经脊髓炎谱系疾病诊断与治疗指南》。其中明确,以AQP4-IgG进行诊断分层,可将NMO分为AQP4-IgG阳性组与阴性组。对于AQP4-IgG阳性组,仅需一项核心临床特征,并且排除其他诊断即可确诊;对于AQP4-IgG阴性组,则需要更多且更加严格的附加条件才可确诊。
多发性硬化症(MS)是另一种炎性脱髓鞘病,与NMO的临床表现相似,在诊断时易与NMO混淆。NMO主要应用免疫抑制剂治疗,而MS以免疫调节治疗为主,所以误诊会给患者造成较大危害。AQP4-IgG可以对MS与NMO进行鉴别诊断,提高临床诊断的准确性。
一、AQP4-IgG检测试剂上市批准情况
英国RSR公司的AQP4-IgG检测试剂先在欧洲上市,RSR公司又通过合作商KRONUS, Inc.使其产品在美国上市。另外,Cosmic Corporation采用RSR公司原材料生产的试剂在日本上市,德国欧蒙公司的AQP4-IgG间接免疫荧光检测试剂盒(IIFT)在欧洲上市。由于我国尚无AQP4-IgG检测试剂上市,在临床实际中,部分患者很难确诊,只能给予脱髓鞘病这一宽泛的临床诊断,甚至有部分患者被误诊为MS,导致接受错误的治疗。2018年,RSR公司向国家药品监管局医疗器械技术审评中心提交了水通道蛋白抗体(AQP4 Ab)测定试剂盒(酶联免疫法)的注册申请。作为诊断罕见病且具有明显临床优势的产品,该产品已进入优先审批程序。
二、AQP4-IgG检测试剂的审评思路
(一)临床试验
作为国内同品种首个产品,因无国内已上市同类试剂,AQP4-IgG检测试剂的临床试验无法与已上市试剂进行比较研究,因此,须通过使用国外已上市产品作为实验室检测方法进行比对,同时结合疾病诊断的临床参考标准进行综合评价,以确定其临床灵敏度和特异度。
由于存在AQP4-IgG阴性的NMO,所以该指标灵敏度存在不足。但作为NMO特有的生物免疫标志物,AQP4-IgG具有高度特异性,相关指南也对其特异性进行了强调。如果特异性差,出现假阳性,则极易造成误诊。本产品采用酶联免疫法,相对较敏感,容易出现假阳性,所以在临床试验中,应入组NMO病例及非NMO病例,非NMO病例应包括临床诊断为MS、其他脱髓鞘病和其他自身免疫病的病例,重点关注其特异度结果。临床试验病例数估算采用诊断试验样本量的估算模型,根据产品临床应用预期的灵敏度及置信区间估算阳性病例数量,根据产品临床应用的特异度及置信区间估算阴性病例数量。临床试验评价指标为置信区间的宽度是否满足期望值。
由于NMO阳性病例难以收集,如果产品临床研究中阳性例数不满足统计学要求,但在已有试验数据可在一定程度上提示产品的灵敏度及置信区间满足临床应用的前提下,可认同其临床试验。同时依据《用于罕见病防治医疗器械注册审查指导原则》,附带条件批准产品上市,要求注册人继续总结产品临床应用情况,并对产品进行上市后评价。
(二)阳性判断值研究
由于该产品具有明显的辅助诊断价值,其阳性判断值研究资料至关重要,因此应采用来源于NMO患者、MS患者、其他脱髓鞘病和其他自身免疫病患者及健康人群的真实样本,对阳性判断值进行充分研究,建议采用受试者工作特征曲线(ROC)的分析方法,研究确定阳性判断值。
(三)定性或定量的考量
由于AQP4-IgG没有公认的标准品或参考品,需通过稀释检测多例阳性样本,对一定吸光度值的样本赋予量值,从而建立稳定的企业参考品和溯源体系,这种溯源方法属于相对定量。在日本和欧盟上市的AQP4-IgG检测试剂为定量检测,在美国上市的AQP4-IgG检测试剂为半定量。目前临床使用中,对于AQP4-IgG浓度的高低是否具有临床意义存在争论,部分观点认为监测AQP4-IgG的浓度对疾病进展和复发预测具有一定的应用价值,但是由于罕见病例数、试剂的可获得性欠缺等诸多因素,这一观点尚未经过充分证实。
包虫病临床诊断及病原体相关游离DNA(cfDNA)简介
一、包虫病简介
包虫病又名棘球蚴病,是一种古老的人畜共患性寄生虫病,主要由人感染棘球绦虫的幼虫所致。该疾病分为囊型包虫病和泡型包虫病两种,分别由细粒棘球蚴和多房棘球蚴感染引起。两种绦虫都必须在哺乳类动物体内寄生才能完成生活史,细粒棘球绦虫的终末宿主一般为犬和狼;多房棘球绦虫的终末宿主一般为狐狸、犬和狼。
近年来,包虫病呈全球性分布,我国包虫病主要流行于西北牧区和半农半牧地区,其中以新疆、西藏、宁夏、甘肃、青海、内蒙古、四川7省最为严重。根据文献报道,我国西部人群包虫病的感染率为3.1%~ 31.5%,患病率为0.5%~5.0%,其中青藏高原部分地区人群患病率为5.0%~10.0%。据2010年原国家卫生和计划生育委员会“防治包虫病行动计划(2010―2015)”,我国西部地区包虫病平均患病率为1.08%,受威胁人口约为6600万,每年造成直接经济损失30亿元。
二、包虫病诊断依据
根据现行包虫病诊断标准,包虫病的临床诊断要综合考虑:流行病学史、临床表现、影像学检查、实验室检查及病原学检查几个方面。
1.流行病学史
该部分主要考虑患者是否有在流行区的居住、工作、旅游或狩猎史,或与犬、牛、羊等家养动物或狐、狼等野生动物及其皮毛的接触史;在非流行区有从事来自流行区的家畜运输、宰杀、畜产品及皮毛加工等暴露史。
2.临床表现
患者主要的临床表现为棘球蚴的囊占位所致的压迫、刺激、囊破裂引起的一系列症状,囊性包虫病可发病全身多个器官,以肝、肺为常见;泡型包虫病原发病灶几乎都位于肝脏。
3.影像学检查
主要为通过B超扫描、X线检查、计算机断层扫描或磁共振检查发现占位性病变。
4.实验室检查
通过酶联免疫吸附试验、间接红细胞吸附试验、PVC膜快速ELISA、免疫印迹技术等方法对包虫病相关的特异性抗体、循环抗原、免疫复合物进行检测。
5.病原学检查
在手术活检材料、切除的病灶或排出物中发现棘球蚴的囊壁、子囊、原头节或头钩。此为包虫病的确诊方法。
三、相关检测产品介绍
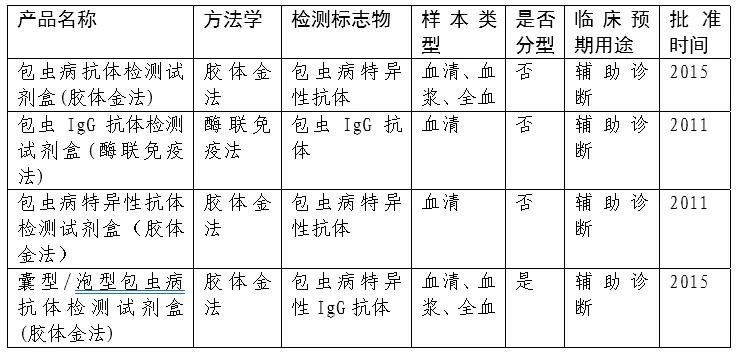
对来源于患者或疑似患者人体样本中的包虫病感染相关生物标志物进行检测是辅助诊断包虫病的重要手段。相关生物标志物包括特异性抗体、循环抗原、免疫复合物、病原体核酸等。我国已批准四个包虫病抗体检测试剂,产品信息见表1,已批准产品均采用免疫学方法对样本中的病原体抗体进行检测,免疫学检测有其自身的局限性,一方面,由于细粒棘球蚴和多房棘球蚴两种病原体自身抗原存在交叉的特点,导致应用抗体检测来区分囊型/泡型效果欠佳;另一方面,抗体检测无法区分现症感染与既往感染。随着检测技术的进步,病原体核酸及其游离DNA(cfDNA)检测为感染的辅助诊断提供新的思路。目前,国际上尚无包虫病核酸检测相关的产品,我国已有细粒棘球蚴和多房棘球蚴核酸检测试剂盒(PCR-荧光探针法)产品获批进入国家药监局创新医疗器械特别审查程序,该产品临床应用的安全有效性仍然在验证过程中。
表1 我国已批准的包虫病检测试剂
四、病原体相关游离DNA(cfDNA)简介
近年来,随着对寄生虫血浆游离DNA(cfDNA)的研究,cfDNA在寄生虫病的检测中得到了快速发展,目前已有应用cfDNA检测对疟原虫、弓形虫、利士曼原虫、锥虫、血吸虫等寄生虫感染进行辅助诊断的报道。针对寄生虫感染后cfDNA的来源,可能是虫体或虫卵的细胞或碎片在增生、成熟、脱落、腐烂、崩解等过程中主动分泌或被动释放出内部的DNA进入宿主体液循环系统产生;也有研究表明绦虫主动分泌的外泌体是寄生虫感染后患者体内出现虫源DNA的重要原因。基于上述研究,检测样本中细粒棘球蚴和多房棘球蚴两种病原体游离DNA为该疾病的辅助诊断提供了理论基础。
五、检测游离DNA的考量
寄生虫源cfDNA的检测在临床应用中存在一定优势,一方面,可根据特异性的基因序列对两种不同种类的病原体进行分型,从而达到对疾病进行分型的目的;另一方面,患者样本中存在寄生虫DNA在一定程度上可提示患者可能感染相关病原体。然而,作为一个新的标志物,能够应用于临床,在现有的理论基础上,还应进一步研究。一是,应进一步明确患者不同疾病进展阶段血浆游离DNA浓度的变化,并以此为基础,确定该标志的临床预期用途(如:疾病早筛、辅助诊断、鉴别诊断及治疗监测等);二是,应依据标志物拟定的预期用途,进一步对该标志物进行临床验证,临床研究应考虑入组人群、对照方法等,如该标志用于疾病的鉴别诊断,则入组人群应为包虫病疑似人群,其中不同大小占位性病变病例、不同病灶退变(CE4、CE5)病例、不同基因型病例等均应入组,此外还应考虑非包虫病病例的干扰。用于疾病鉴别诊断用途的临床研究其对照方法应为疾病确诊的标准即病原学检查。
六、总结
我国包虫病防控形势严峻,包虫病流行区多为相对偏远、落后的地区,目前包虫病的诊断很大程度上依赖于影像学和病原学,这不仅对临床医生专业技术水平要求较高,还存在一定程度的误诊、漏诊的风险。血浆游离cfDNA检测操作较为简便,结果易于解读,如能按预期应用于临床,将对包虫病的诊断起到积极作用。
乙型肝炎病毒耐药基因检测概述
一、乙型肝炎病毒耐药基因检测的临床意义
WHO相关资料显示,全球感染过乙型肝炎病毒(HBV)的患者超过三分之一,而慢性乙型肝炎患者约有2.4亿,乙肝严重影响着人类的生命健康。HBV是传染性疾病乙型病毒性肝炎的主要病因,感染HBV可引起肝硬化甚至肝细胞癌变。HBV属嗜肝DNA病毒科,为双链DNA病毒,容易发生变异,从而形成不同的基因型。目前已根据HBV核苷酸序列异质性≥8%将其分成A-J 10种基因型。在我国北方以C型为主,南方以B型为主,新疆有出现D型。作为常规抗病毒治疗的核苷(酸)类似药物,拉米夫定(LAM)和阿德福韦酯(ADV)等能够有效抑制HBV的复制,然而长期使用会导致耐药的发生,使治疗效果明显下降。随着分子生物学的快速发展,越来越多的分子生物学技术用于HBV的基因分析,从而为乙肝患者的抗病毒治疗提供科学的依据。尽早检测HBV变异,准确判断拉米夫定和阿德福韦酯等治疗后耐药,指导个性化用药,对于提高乙肝治疗成功率具有很重要的价值。
二、乙型肝炎病毒耐药机制以及常见的突变基因位点
HBV基因型耐药是指乙肝病毒出现某种特定的突变,这些特定的突变点导致HBV耐药。其分子机制是:HBV是一种变异较高的病毒,在复制过程必须经过RNA中间体的逆转录复制步骤,但RNA聚合酶和逆转录酶缺乏校正功能,因此在宿主免疫压力和抗病毒药物的选择压力下,其核酸在少数位点上发生突变是一种常见现象。
HBV对核苷(酸)类似物耐药的产生与HBV聚合酶基因变异有关,并且HBV聚合酶基因变异是多位点的,以C区YMDD(酪氨酸-蛋氨酸-天冬氨酸-天冬氨酸)基序的变异最为重要。拉米夫定(LAM)的主要耐药位点位于C区(rtM204V/I),204位点的蛋氨酸被缬氨酸(V)或异亮氨酸(I)置换,即rtM204V/I。上述突变直接导致病毒对药物的敏感性降低,被称为原发耐药突变。另外部分突变不直接引起病毒对药物敏感性的下降,但可以恢复原发耐药变异病毒受损的复制力,被称为补偿耐药突变,主要有rtV173L、rtL180M(常与rtM204V共同出现)和rtL80I(常与rtM204I共同出现)。比如,HBV双点突变株如rtM204V/rtL180M,其病毒复制能力强于rtM204I的单点突变株,可能与rtL180M(B区)的变异补偿C区YMDD变异株的复制缺陷有关。而另一种代表药物阿德福韦酯(ADV),其耐药变异位点则主要集中于HBV聚合酶基因B区rtA181T和D区rtN236T位点。
三、乙型肝炎病毒耐药基因检测方法
1.PCR产物直接测序:是将HBV基因组的逆转录酶区进行扩增后直接进行测序分析的方法。PCR产物直接测序法可检测已知和可能的未知耐药变异位点,是最常用的基因型耐药检测方法之一。PCR产物直接测序的方法一般作为基因型耐药检测的金标准。该方法的缺点是灵敏性较差,只有当变异株超过HBV准种池的20%时才能被发现。
2.聚合酶链反应-限制性片段长度多态性(PCR-RFLP):该方法具有较强的灵敏性,可检测数量占HBV准种池5%的耐药变异株。但是PCR-RFLP只能检测已知、单一位点的变异,对于少数耐药变异位点监测不失为一种简便、快速且廉价的方法。但随着多种核苷(酸)类似物的相继问世和HBV耐药变异位点的不断出现,该方法将难以胜任多位点变异的检测。
3.反向杂交法:基于该技术的INNO-LiPA方法在国外已获准应用于临床检测,目前INNO-LiPA HBV DR v3 可检测包括拉米夫定(LAM)、替比夫定(LdT)、恩替卡韦(ETV)与阿德福韦酯(ADV)常见耐药位点。该法可检测变异株占HBV准种池5% ~ 10%的样品,故敏感性较好,但其同样只能检测已知位点变异。
4.实时荧光PCR:该方法操作简便,可检测变异发生率低于10%的耐药变异。仅能检测已知位点。
5.基因芯片:亦称DNA芯片、DNA微阵列,可检测已知变异位点。
6.限制性片段质谱多态性技术:该技术是将PCR-RFLP技术与基质辅助激光解吸电离飞行时间质谱技术结合,灵敏度高,能够发现数量不足HBV准种池1%的变异株,但其同样仅能检测已知位点变异,且价格昂贵,很难在临床推广应用。
四、目前已上市产品介绍
我国已批准上市的乙型肝炎病毒耐药基因检测试剂盒较多采用荧光PCR法,主要为国产产品,针对的基因突变位点主要是204位点的YMDD→YIDD(550ATG→ATT及550ATG→ATC)与YMDD→YVDD(550ATG→GTG)。还有较少产品能够检测发生在180位点(rtL180M)的基因突变。
五、小结
由于肝细胞中的共价环状闭合DNA持续存在,使得乙肝患者需要长期用药治疗。抗HBV核苷(酸)类药物主要有拉米夫定(LAM)、替比夫定(LdT)、恩替卡韦(ETV)、阿德福韦酯(ADV)和富马酸替诺福韦酯(TDF),其中前4种已在我国上市。长期使用核苷(酸)类似药物不可避免地导致耐药基因突变,最终病毒恢复复制能力,病情反弹。尽早进行乙肝病毒耐药基因突变的检测,可以及时发现病毒的耐药情况,指导医生合理用药。随着生物检测技术的发展,越来越多的突变位点被发现,为此类产品的技术审评带来挑战。
体外诊断试剂与适用仪器的技术审评情况介绍
一、背景介绍
《体外诊断试剂注册管理办法》规定,对于已获批的试剂增加适用仪器情况,注册人可以采取申请体外诊断试剂产品许可事项变更的形式进行注册申报。区别于注册申请,对于上述适用于变更的情况,注册人可以在不改变产品的设计以及技术原理的情况下,进行设计变更的验证或确认,更加灵活地进行产品的研发和生产。
二、我国的技术审评情况及发展趋势
随着已获批体外诊断试剂增加适用仪器的现象层出不穷, 本着科学审评的原则,参考国际先进监管经验,我们也在工作过程中探索新的审评方式。具体的实施路径是由注册人具体变更情形,从技术分析和风险管理两个角度分析并说明变化部分对产品安全性、有效性可能产生的影响。根据上述分析和考量,自行选择验证或者确认变化部分对产品安全性、有效性影响的研究方法,包括分析性能以及临床性能研究部分,并制定研究方法的选择依据以及研究结果的验收标准。在此基础上制定研究方案,开展对试剂盒和新增适用仪器组合的研究,详细记录研究过程和结果以及得出结论。另外,技术审评鼓励注册人尽可能系统全面的分析和论证各类证据用于支持增加适用仪器的科学合理性。
注册人可以通过基于产品风险的评估,相应地进行设计变更的验证与确认,分析并评价新增加的适用仪器对产品说明书声称的分析性能和临床性能的影响。其中分析性能包括对于试剂盒的性能以及试剂盒和仪器组合性能的评估验证,前者例如准确度、检测限、精密度、线性或溯源性等,后者例如携带或交叉污染、机载试剂和定标液稳定性或钩状效应等。同时应研究确认对于临床性能的影响,例如是否改变预期适用人群的参考区间或者阳性判断值。另外还包括对临床适应症的影响,例如在定性、半定量和定量结果判读更改,样本类型的更改或者增加高灵敏度性能的情况下评价诊断灵敏度和特异性、阴阳性符合率和总符合率等关键临床参数。
根据上述对于试剂盒和仪器组合进行评估的验证或确认研究,在结果显示变更对产品的安全有效性不造成显著影响,并且不引发新的风险或者显著改变现有风险的情况下,注册人即可以产品许可事项变更的形式进行注册申报。
这里值得注意的是,在影响医疗决策制定,也就是显著改变临床性能的情况下,基于对产品临床使用过程中潜在风险的考虑,建议注册人将变更后的试剂盒按照新产品进行注册申报。
三、其他国家的监管情况
对于类似的情况,美国FDA于2003年发布了仪器替代家族政策,并于2017年12月进行了更新。适用范围是已获批的体外诊断试剂产品增加适用仪器的情况,前提是适用仪器已上市或者与已获批的设备属于同一个仪器家族。这里的仪器家族是指具有相同的结构、设计、电气安全和电磁兼容性能以及功能(如检测方法、信号范围和强度以及反应条件)的设备系列;试剂和仪器可以是相同或者不同厂家生产的产品。注册人应当根据检测试剂盒的质量体系要求评估试剂盒和新增适用仪器组合的性能,以确保申报的检测系统具有可接受的性能。此外,试剂盒的注册人负责确保经调整或更改的检测系统能够持续满足设计验收标准。
对于试剂盒和新增适用仪器组合的性能研究,也就是将已获批的检测系统的性能转移到新的检测系统后的评价路径,美国FDA也发布了相关的技术指南性文件,并建议将研究结果纳入PMA(上市前批准)补充文件或新的510(k)(上市前通告)文件。包括定性检测和定量检测产品的转移研究,其中定性检测应评估产品的诊断灵敏度、精密度以及使用临床样本进行比较研究等;定量检测应评估产品的分析灵敏度、精密度、线性范围以及使用临床样本进行方法学比对研究等。
根据上述对于试剂盒和仪器组合进行评估的验证或确认研究,在结果显示变更对产品的安全有效性不造成显著影响,并且不引发新的风险或者显著改变现有风险的情况下,注册人即可考虑将检测试剂盒应用于仪器家族的新成员,并按照质量管理体系的要求进行相应的文件记录或者提交PMA(上市前批准)补充文件申请。
如果发生下属情形之一,美国FDA则要求注册人提交新的510(k)或者PMA申请。第一种情况是体外诊断试剂的技术原理发生改变,包括反应原理(如双抗原夹心法、杂交捕获法),检测模式(如化学发光法、比浊法),测量方法(如终点/速率、定性/定量),信号处理方法,数据获取和解读方式,分析前处理步骤等。第二种情况是主要原材料或关键反应成分发生更换,包括抗原-抗体、酶-底物、结合物、信号标记物、固相载体、引物-探针、目标物分离(如核酸提取)组分等。
全面了解艾滋病病毒检测试剂
相关背景
2019年12月1日是第32个“世界艾滋病日”,我国宣传活动的主题是“社区动员同防艾,健康中国我行动”。目前,我国艾滋病防控任务依然艰巨。
艾滋病的诊断原则是以实验室检测为依据,结合临床表现并参考流行病学资料综合进行。本文对艾滋病病毒检测试剂的主要种类及各自的用途特点进行介绍,帮您全面了解艾滋病病毒检测试剂和检测方法。
艾滋病,即获得性免疫缺陷综合征(acquired immunodef i ci ency syndr ome,AIDS),是由人类免疫缺陷病毒(human i mmunodef i c i ency vi r us,HI V)也就是艾滋病病毒感染引起的,以人体CD4+T淋巴细胞减少为特征的进行性免疫功能缺陷,疾病后期可继发各种机会性感染、恶性肿瘤和中枢神经系统病变等,病死率较高。
目前,艾滋病是威胁我国公众健康的重要公共卫生问题。据中国疾病预防控制中心、联合国艾滋病规划署、世界卫生组织联合评估,我国新发感染者每年在8万例左右,截至2018年底,我国估计存活艾滋病感染者约125万,全人群感染率约9人/万人。
HIV检测试剂根据检测目标物的不同大致分为:抗体检测试剂、抗原检测试剂、核酸检测试剂和HIV基因型耐药检测试剂。通常人们也会根据检测试剂的使用特点、用途、性能等,将其进行归类,如HIV抗体快速检测试剂、HIV自测试剂、HIV抗体筛查试剂、HIV抗体诊断试剂、HIV抗体确证试剂等。检测的样本类型包括血液、尿液、口腔黏膜渗出液等。
一、HIV检测试剂分类:
1.发光类检测试剂
从HIV感染人体到感染者血清中的HIV抗体、抗原或核酸等感染标志物能被检测出之前的时期,称为艾滋病的窗口期。此阶段的感染者具有传染性,但无法被检测手段识别。随着检测手段的迭代更新,艾滋病窗口期被一再缩短。目前市场上的主流艾滋病辅助诊断试剂为第三代发光类检测试剂和第四代发光类检测试剂。第三代试剂检测的是HIV-IgG和HIV-IgM抗体,窗口期3周左右;第四代试剂同时检测HIV抗体和p24抗原,窗口期2周左右。
2.抗体快速检测试剂
目前已有多家生产商生产的快速检测试剂获批上市。此类试剂采用胶体金等免疫层析技术原理,检测受试者血液、口腔黏膜渗出液等样本中的HIV抗体。快速检测试剂一般只需20分钟即可获得结果。然而,快速检测并不等同于早期诊断,并不能更早地发现HIV感染。相反,基于该检测方法的局限性,当采用非血液样本进行检测时,应谨慎可能出现的假阴性及假阳性结果。
3.自测试剂
扩大检测(尽可能多地发现AIDS患者/HIV感染者)是我国预防控制艾滋病的核心策略。由于此疾病的隐匿性,及社会歧视、耻辱感等的存在,高危人群获得上述医学专业实验室检测的覆盖率仍不足。
今年7月,我国首次批准了可用于消费者自测用途的HIV尿液抗体检测试剂。与上述其他检测试剂不同,自测试剂在试剂性能满足要求的基础上,其产品设计更易操作,说明书编写更为通俗,适合无专业背景的人使用。此类试剂在上市前临床试验研究中,对目标用户在医院及家中等可能应用的场景进行自检试验,并进行了消费者对说明书、标签等关键信息的调查问卷试验,确认了无专业背景消费者使用时检测结果的可靠性。
4.HIV抗体确证试验试剂
上述发光类检测试剂、抗体快速检测试剂和抗体自测试剂,都属于抗体初筛试验类试剂范畴,但人们无法仅根据初筛试验结果给出受试者感染HIV的结论。临床中,对初筛阳性人群,需采用包括免疫印迹法(WB),条带/线性免疫试验(RIBA/LIA),间接免疫荧光(IFA)等方法进行复检试验,此类试剂被称为HIV抗体确证试验试剂。
5.HIV核酸检测试剂
患者感染HIV后,病毒在其体内快速复制,当血液中的病毒RNA高于核酸检测试剂检测下限时,HIV核酸检测结果呈阳性,此时可结合流行病学史、临床症状及HIV抗体初筛结果作出判断。目前认为HIV核酸检测的窗口期可缩短至1周左右。
除上述初筛用途外,HIV核酸定量检测试剂还可用于临床管理HIV感染的患者。具体来说,可通过测定基线HIV-1RNA水平,对患者的预后进行评价;或在抗病毒治疗过程中通过测定患者血液中HIV-1RNA水平的变化来监控抗病毒治疗的效果。
6.HIV基因型耐药检测试剂
HIV耐药检测结果可为艾滋病治疗方案的制订和调整提供重要参考。检测结果为HIV耐药,表示该感染者体内病毒可能耐药,同时需密切结合临床情况,充分考虑HIV感染者的依从性,对药物的耐受性及药物代谢吸收等因素进行综合评判。
二、注意事项
HIV感染的高风险人群包括有男男同性性行为者,静脉注射毒品者,与HIV/AIDS患者有性接触者,多性伴人群,怀疑接受过不洁输血、使用过未经严格消毒的针具注射的人等。当以上人群出现发烧、皮疹、疲劳、头痛、腹泻、淋巴结肿大等症状,或怀疑自己可能感染HIV时,可到设置于当地疾病预防控制机构或指定医疗机构的艾滋病自愿咨询检测(VCT)门诊/室,接受免费且保密的咨询和检测,也可自行购买自测抗体检测试剂,进行自我检测。
消费者进行自我检测时应特别注意:由于疾病窗口期的存在、HIV变异或样本处理不当等因素,若检测结果为阴性,并不能排除HIV感染的可能性。如果自我检测距离最近一次高危行为的时间间隔在窗口期内,建议隔一段时间再自行检测,或到当地疾病预防控制机构或医疗机构咨询检测。若自我检测结果呈阳性,也无需过分恐慌,因为自我检测结果仅为初筛结果,并不能判定检测者已感染HIV,此时,应向所在地的疾病预防控制机构或医疗机构进行咨询,并做进一步检测来确诊HIV感染状态。
目前,国家已批准了多个用于HIV感染检测的试剂,用途覆盖艾滋病的诊断、病情监测和用药指导。就使用人群来区分,获批产品包括实验室检测试剂和消费者自测试剂。需注意的是,目前可选择的所有检测试剂都有各自的用途和局限性,使用时应根据用途和行业技术规范,确定HIV感染检测策略。
微卫星不稳定性(MSI)检测概述
一、微卫星不稳定性(MSI)简介
微卫星不稳定性(Microsatellite Instability,MSI),是指由于在DNA复制时插入或缺失突变引起的微卫星(Microsatellite, MS)序列长度改变的现象,常由错配修复功能(Mismatch repair, MMR)缺陷引起。MS序列,是一些短而重复的DNA序列,一般由1~6个核苷酸组成,串联重复排列,常见类型为双碱基CA/GA/GT或单碱基A/T等。MS序列可以位于基因的重要非编码区,也可以位于基因的编码区,多态性分布于整个基因组,个体差异大。
MSI现象于1993年被Jacobs等人在结直肠癌中首次发现。随着针对MSI的研究深入,发现MSI现象不止存在于结直肠癌,在子宫内膜癌、胃癌、肝细胞癌、乳腺癌等实体瘤中均有发生。
二、MSI检测方法
目前检测癌细胞中的MSI时,既可以通过检测MMR基因缺失来确定是否发生MSI,如依赖于免疫组化技术的蛋白水平检测,也可以直接检测MSI的序列变化,如PCR(聚合酶链反应)检测等的分子水平检测。
(一)免疫组化方法(Immunohistochemistry, IHC)
常见的方法为采用免疫组织化学方法检测肿瘤组织中错配修复基因MLH1、MSH2、MSH6及PMS2的表达。任何一项错配修复基因表达缺失,被定义为MSI,否则即为微卫星稳定(MSS)。其中一个表达缺失则称为低度微卫星不稳定(MSI-L),2种或2种以上蛋白表达缺失为高度微卫星不稳定(MSI-H)。
此方法检测MSI相对较简单,成本较低。但存在一些问题,如使用抗体的不一致、病理医生的阅片标准不一致等。
(二)分子水平的检测
1.聚合酶链反应(polymerase chain reaction, PCR)技术
目前主要采用多重荧光PCR结合毛细管电泳的方法。通过PCR方法检测特异的微卫星重复序列扩增判定MSI状态,比较肿瘤患者的标本组织与正常组织的位点突变情况。其比较的位点为美国国家癌症研究所(NCI)推荐的5个微卫星位点:BAT25、BAT26、D5S346、D2S123及D17S250。其中≥2个位点发生改变判定为高度微卫星不稳定(MSI-H),仅1个位点发生改变判定为低度微卫星不稳定(MSI-L),无位点改变判定为微卫星稳定(MSS)。有研究认为,二核苷酸重复序列的特异性和灵敏度比单核苷酸重复序列要低,因此关于最适合MSI检测的位点仍存在争议。Bacher等对266个微卫星位点(其中包括单核苷酸、二核苷酸、四核苷酸以及五核苷酸微卫星位点)检测的敏感性及准确性进行评估,提出了Promega分析系统,该系统使用五个单核苷酸微卫星位点(BAT-25, BAT-26, NR-21, NR-24和MONO-27)及两个五核苷酸微卫星位点(Penta C和Penta D)检测MSI。
大量的实验证实,MMR免疫组化检测结果与MS的PCR检测结果有高度关联性,灵敏度92%,特异性可达100%。
该方法目前是检测MSI的方法学“金标准”,但操作过程复杂,花费较高,且在结果判断中会碰到以下问题,如荧光的过强或过少、非特异性峰、不显著的峰大小改变,杂合性缺失等。
2.新一代测序方法(Next Generation Sequencing, NGS)
NGS又称为第二代测序技术,是一种高通量测序技术,能一次性对几十万到几百万条基因分子进行序列测定。目前NGS方法已经成为检测MSI的新工具,其最大优势是可以实现多位点高通量检测。纪念斯隆・凯特琳癌症研究中心(Memorial Sloan Kettering Cancer Center,MSK)的一项研究使用NGS方法对12,288例实体瘤病人进行检测,判定MSI状态,并用MSI-PCR/MMR-IHC进行了验证。实验证明, NGS方法较MSI-PCR方法具有更高的敏感性。
三、MSI检测的临床意义
目前,有关MSI检测的临床意义有如下观点:
(一)Lynch综合征的筛查
Lynch综合征,亦称遗传非息肉病性结直肠癌(HNPCC),是一种呈家族遗传倾向的常染色体显性遗传病,由于MMR基因发生胚系突变所致,约占所有结直肠癌(Colorectal carcinoma, CRC)3%~5%。Lynch综合征最突出的特点之一是携带者本人或家族成员可发生CRC及其他多种Lynch相关肿瘤。MSI是Lynch综合征的特征性分子,约90%以上表现出MSI。同时并不是所有的MSI均是Lynch综合征,在散发性CRC中也存在10%~15%的MSI。
(二)MSI与结直肠癌的预后
临床研究证实,MSI与结直肠癌的预后有着密切的关系。MSI-H结直肠癌患者相比MSS患者具有显著的生存优势,临床表现较差,但预后更好。研究证实,针对Ⅱ/Ⅲ期结直肠癌患者,MSI-H患者的总生存期及无病生存期明显延长。美国国家综合癌症网络(National Comprehensive Cancer Network,NCCN)发布的结直肠癌指南,建议MSI检测应在所有结直肠癌史的病人中进行。
(三)MSI与指导用药
研究证实,MSI-H的结直肠癌Ⅱ期患者不能在氟尿嘧啶(5-FU)治疗中获益。同时因MSI-H的晚期结直肠癌患者通常具有PD-1及PDL-1等的高表达,可通过对PD-1/PD-L1的靶向抑制,促使机体免疫系统攻击和杀灭肿瘤细胞。目前美国FDA已批准PD-1免疫治疗单抗pembrolizumab、ipilimumab和nivolumab用于具有错配修复缺陷(dMMR)/MSI-H表型的转移性结直肠癌(mCRC)的末线治疗。除了mCRC的治疗外,2017年5月,美国FDA批准了PD-1抗体pembrolizumab用于治疗成人和儿童具有dMMR/MSI-H表型的没有其他治疗选择的,不可切除或转移性实体瘤患者。
《中国结直肠癌诊疗指南》(2010版)就明确了“Ⅱ期结直肠癌,有条件者建议检测组织标本MMR或MSI,如为dMMR或MSI-H,不推荐氟尿嘧啶类药物的单药辅助化疗”。在《中国结直肠癌诊疗指南》(2017版)的组织病理学检查部分,增加了MMR或MSI的检测作为复发或转移性结直肠癌的推荐检测方法,“确定为复发或转移性结直肠癌时,推荐检测肿瘤组织K-ras及N-ras基因、BRAF基因、错配修复蛋白表达或微卫星状态及其他相关基因状态以指导进一步治疗。”
四、相关检测产品介绍
目前国际上已上市的产品有:
(一)MSI-IVD Kit (FALCO)
日本FALCO公司的MSI-IVD Kit已获得日本厚生劳动省的批准上市。该试剂盒采用PCR技术检测癌组织中提取的基因组DNA中的MSI状态,检测7个微卫星位点(五个单核苷酸微卫星位点BAT-25, BAT-26, NR-21, NR-24和MONO-27及两个五核苷酸微卫星位点Penta C和Penta D)。该产品由日本FALCO公司与Promega公司合作开发,作为伴随诊断试剂,用于指导PD-1抗体pembrolizumab在局部晚期或转移性癌症患者中的应用。
(二)VENTANA MMR IHC panel
该产品采用IHC方法检测经福尔马林固定石蜡包埋的结直肠癌组织切片中的四种MMR蛋白MLH1、MSH2、MSH6、PMS2,及BRAF V600E
突变蛋白。用于Lynch综合征的辅助诊断,同时检测BRAF V600E突变蛋白用于散发性CRC及Lynch综合征的辅助鉴别诊断。此产品通过De Novo途径在美国上市。
(三)MSK-IMPACTTM
2017年11月16日,美国FDA批准纪念斯隆・凯特琳癌症研究中心(Memorial Sloan Kettering Cancer Center,简称MSK)基于二代测序技术的癌症基因检测分析平台MSK-IMPACTTM。该产品能够一次对病人肿瘤468个基因的突变进行检测,并且可以检测MSI基因组特征。该产品仅限于在MSK中进行使用,其检测结果不与任何药物联用。该产品的MSI的检测性能通过对10,900名患有66种不同类型晚期实体瘤患者的研究进行确定。
(四)FoundationOne CDx (F1CDx)
2017年11月30日,美国FDA批准了Foundation Medicine公司针对多种实体瘤的二代测序的体外检测产品―FoundationOne CDx(F1CDx)。该产品仅允许在Foundation Medicine公司进行检测。这款产品除了可检测5种肿瘤中的324个基因的突变,还可以检测MSI和肿瘤突变负荷(TMB)两个基因组特征,是FDA批准的首款获得突破性认定的癌症NGS体外诊断检测产品。
五、总结
随着研究的进一步深入,MSI在肿瘤的发生机制中所起的作用越来越被人们所关注。虽然近年来MSI的检测在临床应用方面被日趋关注,但是国内还未有注册获批的产品。2018年已有MSI检测试剂盒通过国家药品监督管理局的创新医疗器械特别审查程序,该产品采用荧光PCR-毛细管电泳法。
如何判定一个体外诊断试剂是否属于防治罕见病相关产品
体外诊断试剂产品是否属于防治罕见病相关产品,应依据《罕见病防治医疗器械注册审查指导原则》(2018年第101号)、《关于公布第一批罕见病目录的通知》(国卫医发〔2018〕10号)及《国家卫生健康委办公厅关于印发罕见病诊疗指南(2019年版)的通知》(国卫办医函〔2019〕198号)等文件判定。如申报产品临床适用症为第一批罕见病目录中的疾病,且依据罕见病诊疗指南(2019年版),该疾病的诊疗流程中需进行申报产品对应的检测项目的检测,则该产品可认定为防治罕见病相关产品。对于申报产品检测项目为新研发的生物标志物,应明确产品预期用途及其与相关罕见病诊疗的关系,从而判定其是否属于防治罕见病的产品
血气检测类产品适用的样本类型是什么
临床应用中,血气检测类产品(包括仪器和试剂)的样本类型一般为动脉全血。审评过程中依据注册申报资料的验证内容,一般将适用样本类型明确为动脉全血。如果产品适用于检测静脉全血、毛细血管全血等其他样本类型,亦应进行充分验证。
体外诊断试剂的配套质控品有何要求
体外诊断试剂的配套质控品用于对检测系统进行质量控制。申请人应对申报试剂检测质控品的预期结果(靶值和靶值范围)进行验证,并将经验证的配套质控品在试剂说明书中予以明确。未经验证的“第三方质控品”、“其他商用质控品”等表述不应出现在产品说明书中。
体外诊断试剂临床试验中,能否采用境外已上市同类产品作为对比试剂
体外诊断试剂的临床试验一般采用试验用体外诊断试剂与临床参考标准和/或已上市同类产品进行比较研究的方法,评价试验用体外诊断试剂的临床性能,为该产品安全、有效性的确认提供科学有效的临床证据。其中,“已上市同类产品”指的是境内批准上市的同类产品。
对于目前临床上没有可参照的临床参考标准或现有临床参考标准不能全面评价产品临床性能的情况,临床试验设计时,在确认被测物临床检测意义的前提下,还应依据现有临床实践和理论基础选择适当的实验室方法进行检测性能评价,例如:与临床公认的、标准化的实验室参考方法进行对比试验。如果有境外已批准上市的同类产品,与试验用体外诊断试剂具有相同的预期用途,且该产品经过了充分的性能验证和确认,实验室检测过程中可实现良好的质量控制,并被临床广泛认可能够用于相关标志物检测,则该产品亦可作为实验室检测方法用作对比方法。
eRPS系统注册申报申请表中IVD产品分类编码应如何选择
按照《关于实施医疗器械注册电子申报的公告》(2019年第46号)的要求,自2019年6月24日,正式启用eRPS系统。eRPS系统中,体外诊断试剂申请表分类编码一栏除固定6840外,增加了对6840细化的产品类别名称,申请人/注册人可在下拉菜单中选择。以下就该细化的分类编码的选择逐一说明:
1、23-001:与中华人民共和国传染病防治法所述疾病相关的病原体检测试剂;
此类产品仅包括《中华人民共和国传染病防治法》中所规定的甲类、乙类和丙类传染病相关的病原体检测试剂。
2、23-002:除23-001外其它致病性病原体抗原、抗体以及核酸等相关的检测试剂;
本类产品包括除《中华人民共和国传染病防治法》中所规定的甲类、乙类和丙类传染病以外的其他致病性病原体相关的检测试剂,如幽门螺杆菌、沙眼衣原体、白色念珠菌、人乳头瘤病毒、EB病毒、单纯疱疹病毒等。
3、23-003:与血型、组织配型相关的检测试剂;
本类产品指ABO、Rh等血型检测试剂、血小板抗体、抗人球蛋白、红细胞抗体检测试剂以及HLA-DNA分型检测试剂等。
4、23-004:与人类基因、遗传性疾病相关的检测试剂;
如染色体非整倍体(DNA)、CYP2C19基因等人类基因相关的检测试剂以及遗传性耳聋基因检测、苯丙氨酸检测等遗传性疾病相关的检测试剂。
5、23-005:与免疫组化、原位杂交、流式细胞分析仪配套相关检测试剂;
指采用免疫组化、原位杂交、流式技术的相关检测试剂,且依据《总局关于过敏原类、流式细胞仪配套用、免疫组化和原位杂交类体外诊断试剂产品属性及类别调整的通告》(2017年第226号)按照第二类和第三类体外诊断试剂管理的产品;
6、23-006:除列入免临床目录的肿瘤标志物以外的其它与肿瘤相关的检测试剂;
本类产品是指除列入免临床目录的肿瘤标志物以外的其他肿瘤相关检测试剂,如甲基化检测等。
7、23-007:列入免临床目录的肿瘤标志物相关的检测试剂;
本类产品仅包括已列入免临床目录的肿瘤标志物相关的检测试剂,如甲胎蛋白(AFP)、癌胚抗原(CEA)等。
8、 24-001:用于蛋白质检测的试剂;
24-002:用于糖类检测的试剂;
24-003:用于激素检测的试剂;
24-004:用于酶类检测的试剂;
24-005:用于酯类检测的试剂;
24-006:用于维生素检测的试剂;
24-007:用于无机离子检测的试剂;
24-008:用于药物及药物代谢物检测的试剂;
24-009:用于自身抗体检测的试剂;
24-010:用于变态反应(过敏原)检测的试剂;
24-011:与麻醉药品、精神药品、医疗用毒性药品检测相关的试剂;
24-012:用于其他生理、生化或者免疫功能指标检测的试剂;
24-013:用于微生物鉴别或者药敏试验的试剂;
自24-001至24-013,与《6840体外诊断试剂分类子目录》(2013版)一致。
以上细分的IVD分类编码对于申报资料提交至eRPS系统后能否进入正确的分配路径至关重要,申请人/注册人应重视并正确进行选择,如出现选择错误的情况,请于立卷审查环节发补后进行更正。
体外诊断试剂产品技术要求附录中主要原材料的供应商该如何填写
体外诊断试剂产品技术要求附录中要求标注主要原材料的来源,如为外购应写明供应商。此处的供应商应为原材料的生产商,而不是经销商或代理商。相应的,注册变更情形中主要原材料的供应商的变更是指原材料的生产商发生变化的情形。
体外诊断试剂临床试验中如采用核酸序列测定、GC-MS/MS等实验室检测参考方法作为对比方法进行比较研究,是否可以委托测试
对于某些目前临床上尚不存在明确的临床诊断“金标准”,亦无可比的同类产品上市的体外诊断试剂,临床试验研究者应依据现有临床实践和理论基础,建立合理的方法,进行比较研究。对于部分体外诊断试剂,临床试验中采用核酸序列测定、GC-MS/MS等实验室检测参考方法作为对比方法进行比较研究,这些方法非临床常规检测技术,需要专门的设备仪器和试验条件,且临床试验机构可能不具备相关检测条件。对于此类情况,申请人应尽可能选择具备相应条件的临床试验机构开展临床试验,确无检测条件的部分临床试验机构可将此部分测试委托给专门的测序机构、具备一定检测资质的实验室进行检测,并对检测结果进行认可。应提交临床试验机构与受委托机构的委托证明文件,并评价对比方法的方法学研究和整体质量。不得委托申请人的实验室进行相关测试。
新研制试剂的配套专用仪器尚未取得注册证,是否可以申请试剂注册
对于新研制体外诊断试剂及其配套专用仪器,由于分属不同的法规管理,因此需分别提交注册申请。但试剂及其专用仪器检测性能的验证和确认是密不可分的整体验证过程,因此,在试剂和仪器均已定型的情况下,并不限定试剂和其配套专用仪器的上市顺序。但试剂注册申请时,应能够确保配套仪器及检测系统定型,如使用非本企业生产的仪器,则所使用配套仪器应已作为医疗器械在中国境内上市,并能够对其在配套仪器上的性能进行全面验证和确认。
体外诊断试剂临床试验中能否使用冻存样本
体外诊断试剂产品临床试验中需使用符合入组标准的病例样本进行试验,在具体样本入组时除注意病例的选择外(此部分内容见共性问题“关于体外诊断试剂临床试验入组病例样本的常见问题”),还应注意样本的保存条件。原则上,临床试验样本的使用应最大可能与试剂临床使用过程中样本的状态一致,如临床使用状态为新鲜采集后检测,则应考虑使用新鲜采集的样本进行临床试验。如临床使用过程中可能存在样本保存过程(如一定条件下冻存),且说明书中样本保存条件明确了相应的样本保存条件及有效期,则临床试验中亦可纳入部分相应保存条件下的样本。无论使用新鲜采集样本还是冻存样本,均应确保入组样本的保存条件和期限符合相应产品样本保存的要求。
体外诊断试剂产品说明书“标识的解释”项涉及修改如何执行
按照《总局办公厅关于体外诊断试剂说明书文字性变更有关问题的通知》(食药监办械管[2016]117号),体外诊断试剂说明书信息性内容的文字性变化可由注册人自行修改,其中包括体外诊断试剂说明书“标识的解释”项目,因注册人按照YY/T0466系列标准完善体外诊断试剂说明书中相应标识的解释内容,导致该项内容变化,但不涉及其他需办理许可事项变更的情况,注册人应自行修改。此处YY/T0466系列标准可为YY/T0466系列标准或其对应的ISO 15223标准。如注册人自行修改,应在延续注册时予以说明。
邮政编码:200052 电话:021-63800152 传真:021-63800151 京ICP备15010734号-10 技术:网至普网站建设
 扫描下载
扫描下载 扫描下载
扫描下载 扫描关注
扫描关注 扫描关注
扫描关注 扫描关注
扫描关注 扫描关注
扫描关注